


Jarosław Króliczewski
Wydział Biotechnologii, Uniwersytet Wrocławski
strony wersji drukowanej: 4-23

Chromatograf FPLC (www.tech-lab.pl).
Na przestrzeni ostatnich lat zaobserwować można stały rozwój technik i metod umożliwiających oczyszczanie białek. Ma to istotny wpływ na wiele dziedzin nauki, a w konsekwencji przekłada się na stały postęp aplikacyjny i zastosowanie przemysłowe. Współczesna aparatura wykorzystywana w procesie izolacji i oczyszczania białek jest coraz bardziej wydajna, umożliwia dokładne monitorowanie procesu oczyszczania, kontrolę jego parametrów, szybkości itd. Niezbędnym uzupełnieniem wymienionych procesów jest stosowanie odpowiednich technik separacyjnych opartych na metodach chromatograficznych.
strony wersji drukowanej: 4-23
Chromatograf FPLC (www.tech-lab.pl).
Na przestrzeni ostatnich lat zaobserwować można stały rozwój technik i metod umożliwiających oczyszczanie białek. Ma to istotny wpływ na wiele dziedzin nauki, a w konsekwencji przekłada się na stały postęp aplikacyjny i zastosowanie przemysłowe. Współczesna aparatura wykorzystywana w procesie izolacji i oczyszczania białek jest coraz bardziej wydajna, umożliwia dokładne monitorowanie procesu oczyszczania, kontrolę jego parametrów, szybkości itd. Niezbędnym uzupełnieniem wymienionych procesów jest stosowanie odpowiednich technik separacyjnych opartych na metodach chromatograficznych.
Stopień oczyszczania białek w dużej mierze zależy od celu do jakiego będą później wykorzystane. Ogólnie możemy powiedzieć, że celem procesu oczyszczania, jest nie tylko usuwanie niepożądanych substancji zanieczyszczających, ale również uzyskanie oczekiwanego stężenia białka i przeniesienie go do środowiska, w którym jest stabilne. Wysoka czystość białek jest pożądana w badaniach strukturalnych a więc w krystalografii, NMR a także w zastosowaniach terapeutycznych w medycynie i weterynarii. W badaniach biochemicznych i molekularnych polegających na określeniu właściwości fizykochemicznych dopuszczalna jest mniejsza czystość. Idealny stopień czystości dla białka to oczywiście 100 %, jednakże za bardzo dobry stopień oczyszczenia preparatu przyjmuje się stan, gdy zanieczyszczenia nie przekraczają 5 %. Oczyszczanie białka jest procesem złożonym, uzależnionym od kilku czynników: źródła białka, lokalizacji komórkowej, budowy białka, jego ilości w naturalnym miejscu występowania itd. Proces oczyszczania zależy również od tego,czy otrzymywane białko ma być uzyskane w formie natywnej, czy jest ono wielodomenowe, bądź czy posiada kofaktory itd.
 Dzięki współczesnym metodom biologii molekularnej uzyskiwanie białek rekombinowanych w większych ilościach jest w znacznym stopniu ułatwione. Z drugiej strony ich oczyszczanie często wymaga zastosowania innych metod niż w przypadku naturalnego źródła ich pochodzenia.
Dzięki współczesnym metodom biologii molekularnej uzyskiwanie białek rekombinowanych w większych ilościach jest w znacznym stopniu ułatwione. Z drugiej strony ich oczyszczanie często wymaga zastosowania innych metod niż w przypadku naturalnego źródła ich pochodzenia. Jedną z bardzo istotnych technik stosowanych do oczyszczania cząsteczek aktywnych biologicznie takich jak białka i kwasy nukleinowe są techniki chromatograficzne. Definiując chromatografię można powiedzieć, że jest to metoda rozdzielania mieszanin, w której rozdzielane składniki ulegają podziałowi pomiędzy dwie fazy, z których jedna jest fazą nieruchomą (stacjonarną), a druga fazą ruchomą (mobilną) układu chromatograficznego. Rolę fazy stacjonarnej może pełnić ciało stałe, ciecz na nośniku lub żel. Fazą ruchomą mogą być: gaz bądź ciecz lub substancja w stanie nadkrytycznym (Supercritical Fluid Chromatography, SFC). W związku z tym chromatografię można podzielić na gazową, cieczową oraz chromatografię z fazą ruchomą w stanie nadkrytycznym, nazywaną fluidalną bądź nadkrytyczną.
Przed rozpoczęciem oczyszczania chromatograficznego rozdzielane białko należy przeprowadzić do roztworu. Istotnym z punktu widzenia wydajnego oczyszczania aktywnego biologicznie materiału jest odpowiedni dobór warunków ekstrakcji. Pierwszym etapem oczyszczania jest izolacja białek z materiału biologicznego z wykorzystaniem metod fizycznych i chemicznych takich jak:
- homogenizacja,
- sonikacja (ultradźwięki),
- liza osmotyczna,
- działanie wysokich ciśnień (prasa Frencha),
- zastosowanie detergentów do uwolnienia białek z błony,
- zamrażanie-rozmrażanie.
Kolejne etapy oczyszczania białek związane są z właściwościami fizykochemicznymi, środowiskiem z którego je wyizolowano, oraz stopniem zanieczyszczenia preparatu. Parametry te mogą spowodować, że ilość otrzymanego białka będzie bardzo niska, może być ono zdegradowane lub utraci swoją aktywność. Podczas oczyszczania może dojść do powstawania agregatów co także może wpłynąć na wydajność separacji danego białka.
Postępowanie w przypadku oczyszczania białek metodami chromatograficznymi zależne jest od kilku czynników, które mogą wpływać na proces oczyszczania i stabilizacji białek:
- Związki redukujące, takie jak ditiotreitol (DTT) czy β-merkaptoetanol zapobiegają utlenianiu grup tiolowych,
- Odczyn pH środowiska. Rozpuszczalność białek w punkcie izoelektrycznym maleje prowadząc do ich agregacji,
- Odporność na czynniki chaotropowe (mocznik, chlorowodorek guanidyny)
- Siła jonowa środowiska,
- Stabilność temperaturowa,
- Dodatek chelatorów (EDTA-Na2),
- Obecność proteaz w preparacie (stosowanie inhibitorów, PMSF),
- Znajomość właściwości białka, mająca bezpośredni wpływ na dobór techniki separacji:
- masa cząsteczkowa (sączenie molekularne, GFC),
- ładunek powierzchniowy białka (w celu ustalenia warunków chromatografii jonowymiennej, IEC),
- powinowactwo (wybór ligandu do chromatografii powinowactwa, AC),
- hydrofobowość (wybór medium do chromatografii hydrofobowej, HIC).
Jedną z najprostszych metod podczyszczania białek jest precypitacja, polegająca na selektywnym wytrącaniu białek z roztworu (Rysunek 1), przy zastosowaniu czynnika precypitującego takiego jak siarczanu amonu (AmSO4) lub glikolu polietylenowego (PEG). Ten drugi stosowany jest do oczyszczania immunoglobulin.
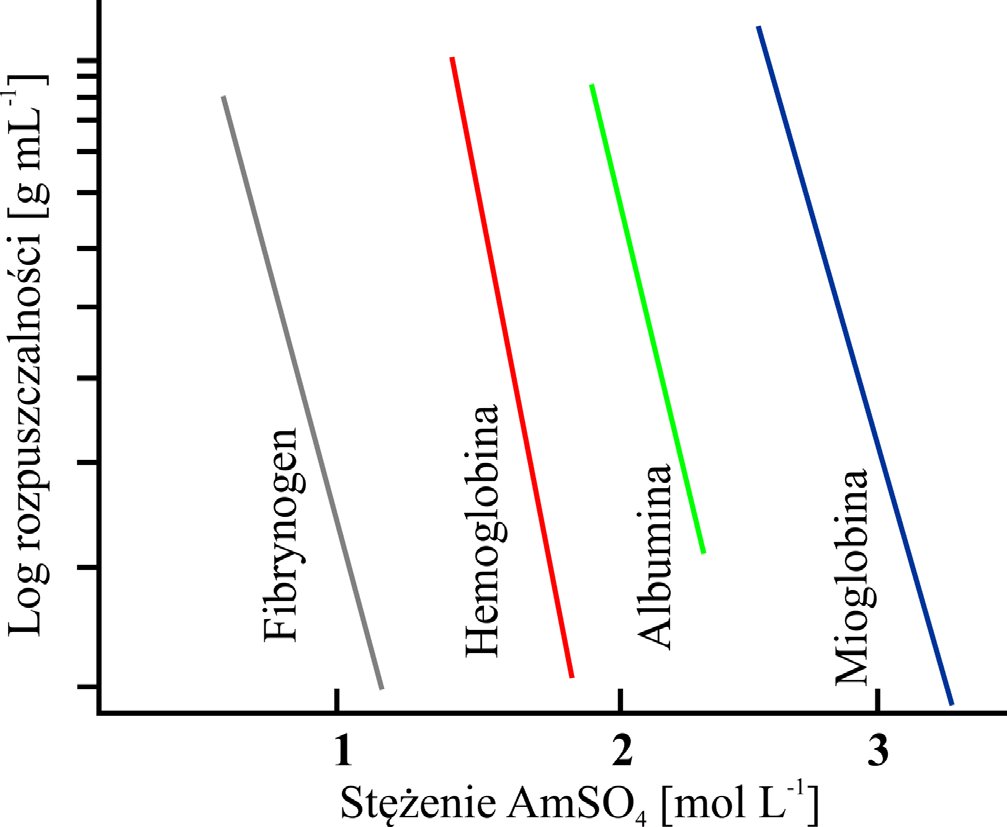
Rysunek 1. Schemat selektywnej precypitacji białek.
W obecności dużego stężenia AmSO4 rozpuszczalność większości białek maleje.
Wytrącanie białek z roztworu związane jest ze zwiększoną ekspresją rejonów hydrofobowych w wyniku usuwania otoczki hydratacyjnej co wywołuje zwiększoną tendencję białek do tworzenia agregatów i wypadania z roztworu. Metoda ta nie jest pozbawiona wad, do których należą mała selektywność i ograniczenia w przypadku niskiego stężenia białek. Większość białek wytrąca się w granicach 40-80 % nasycenia soli (Tabela 1).
Tabela 1. Zakres wysalania AmSO4.
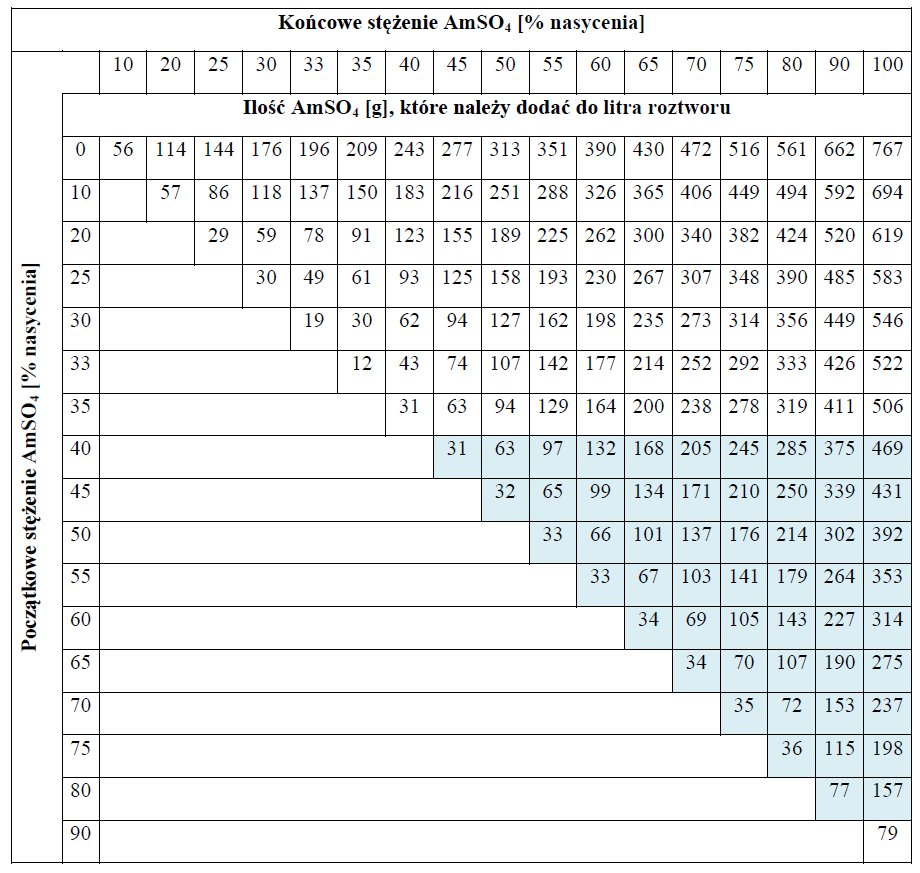
Aby wytrącanie za pomocą AmSO4 było skuteczne, wymagane jest osiągnięcie minimalnego całkowitego stężenia białka 1 mg . mL-1 oraz białka będącego przedmiotem zainteresowania 0,1 mg . mL-1. W przypadku dużych objętości preparatu przed wytrącaniem białek za pomocąAmSO4 zalecane jest zagęszczenie metodą TFF (ultra/diafiltracja).
Przygotowanie próbki przed rozdziałem chromatograficznym
Dobrze przygotowana próbka to połowa sukcesu chromatograficznego oczyszczania białek. Zanieczyszczenia w ekstraktach komórkowych mogą zawierać rozmaite makrocząsteczki (kwasy nukleinowe, micele lipidowe, polisacharydy i inne białka). Mogą one utrudnić lub uniemożliwić rozdział chromatograficzny, a nawet w skrajnych przypadkach zniszczyć filtry chromatografu, zablokować przepływ przez kolumnę lub sprawić że stanie się ona bezużyteczna. Dlatego bardzo ważne jest odpowiednie przygotowanie próbki przed rozpoczęciem oczyszczania chromatograficznego. W pierwszej kolejności należy usunąć wszystkie pozostałości mogące utrudnić rozdzielanie. Najprostszą stosowaną w tym celu metodą jest odwirowanie próbki przed nałożeniem na kolumnę lub przepuszczenie przez filtr o średnicy porów nie większej niż 0,45 µm. Z praktycznego punktu widzenia stosowanie tzw. przedkolumn lub filtrów ochronnych w układach chromatograficznych w celu wyeliminowania skutków tego typu zanieczyszczeń przynosi niewielkie rezultaty. Biorąc pod uwagę aspekt ekonomiczny jest to sposób mało wydajny, ze względu na częste zapychanie filtrów przez różnego typu zanieczyszczenia.
Do problemów związanych z preparatami białkowymi zaliczyć można również ich zwiększoną lepkość związana z zanieczyszczeniem przez kwasy nukleinowe, proteoglikany i polisacharydy. Aby usunąć DNA i RNA najczęściej przeprowadza się preinkubację próbki z nukleazami lub wysalanie siarczanem protaminy (końcowe stężenie 0,01 %), który wykazuje duże powinowactwo do kwasów nukleinowych. Nadmiar polisacharydów możliwy jest do usunięcia dzięki wykorzystaniu np. kolumny wypełnionej ziemią okrzemkową (Celite) lub stosując glukozydazy.
Przy pracy z białkami odpornymi na niskie pH (np. białka fuzyjne z GFP) możliwe jest zastosowanie precypitacji kwasowej (obniżenie pH poniżej 4,6; 4 °C), w której większość zanieczyszczeń ulega wytrąceniu.
Dalsze etapy oczyszczania uzależnione są już od właściwości fizykochemicznych preparatu zawierającego oczyszczane białko i samego białka. Jeśli preparat zawiera niskie stężenie soli, a objętość próby nie jest istotna to możemy zastosować chromatografię jonowymienną.
Chromatografia jonowymienna (IEC)
Chromatografia IEC jest jedną z najczęściej stosowanych technik oczyszczania naładowanych biomolekuł (białka, kwasy nukleinowe, polipeptydy). Duża popularność tej techniki związana jest z prostotą działania, łatwością kontrolowania etapu rozdzielania oraz niskim zużyciem wypełnienia kolumny chromatograficznej, które w łatwy sposób może być wielokrotnie regenerowane. Główną wadą chromatografii IEC jest niestety ograniczona selektywność.
Istnieją dwa główne rodzaje chromatografii IEC: anionowymienna (dla ujemnie naładowanych białek) i kationowymienna (dla dodatnio naładowanych białek). Najczęściej, mieszaniny białek nanoszone są na kolumnę jonowymienną w buforze o niskim stężeniu soli i pH buforu faworyzującym interesujące białko o dodatnim (cation exchange) lub ujemnym (anion exchange) ładunku wypadkowym (Rysunek 2). Oczywiście można stosować odmienne podejście i wykorzystać jonowymieniacz do wiązania pozostałych białek. Wtedyoczyszczane białko wypłynie we frakcji niezwiązanych białek. Co ciekawe, stosując jonowymieniacz możemy zagęścić białko. Należy wtedy użyć buforu o niskim stężeniu (1-10 mM) i pH niższym lub wyższym o jedną jednostkę od wartości pI (punkt izoelektryczny).
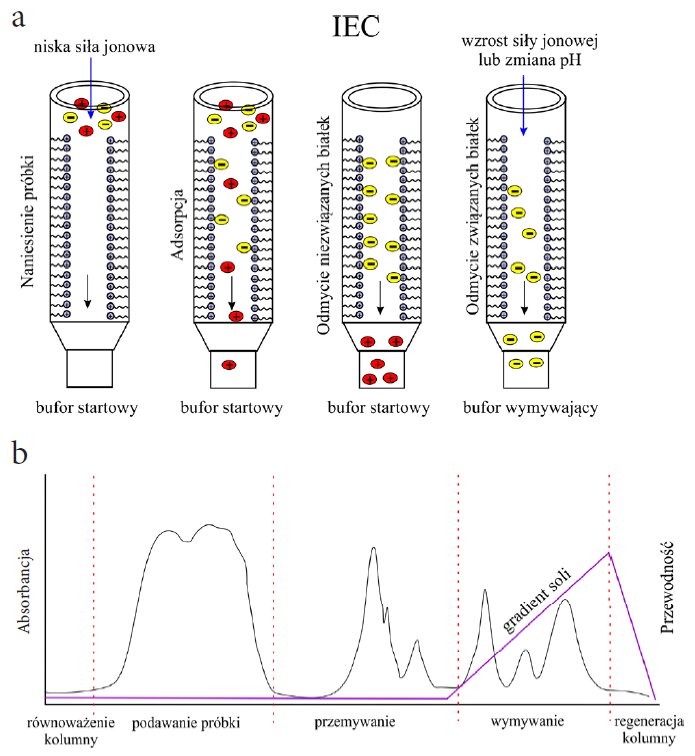
Rysunek 2. Schemat działania chromatografii IEC. (A) schemat działania jonowymieniacza; (B) chromatogram otrzymany w wyniku analizy białek z wykorzystaniem chromatografii IEC.
Białka to złożone amfolity, które posiadają zarówno dodatnie, jak i ujemne ładunki, a ich pI zależy od stosunku jonizacji reszt aminokwasowych (pH > pI ujemny ładunek wypadkowy; pH < pI dodatni ładunek wypadkowy). Ładunki dodatnie pochodzą głównie od argininy, lizyny oraz histydyny i zależą od pH otaczającego buforu. Każdy wolny N-koniec aminy także dostarcza ładunek dodatni w pH poniżej 8. Ujemne ładunki białka zasadniczo pochodzą od kwasów asparaginowego i glutaminowego oraz wolnej grupy karboksylowej na C-końcu. Praktycznie wszystkie te grupy są jonizowane przy pH powyżej 6. Przy wyższych wartościach pH (> 8) jonizacji ulega także cysteina. Naładowane aminokwasy przeważnie zlokalizowane są na powierzchni białka, wyjątek stanowią metaloproteiny.
Niewielkie zmiany ładunku umożliwiają oddzielenie nie tylko całkowicie różnych białek od siebie. Nowoczesna chromatografia jonowymienna zdolna jest do oddzielenia izoform tego samego białka, które różnią się pojedynczym ładunkiem zlokalizowanym na powierzchni białka. Z tego powodu chromatografia IEC często stosowana jest na końcu procesu chromatograficznego oczyszczania białek. Nie zawsze wiązanie białka z jonowymieniaczem zależy od całkowitego ładunku białka. Czasami lokalny ładunek wypadkowy wpływa na oddziaływanie z nośnikiem (Rysunek 3).
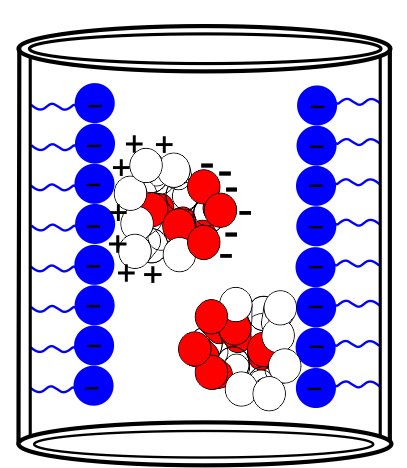
Rysunek 3. Lokalne oddziaływanie pomiędzy ładunkiem nośnika a ukierunkowanym ładunkiem białka.
Korzystając z chromatografii IEC należy pamiętać, że pH mikrośrodowiska wymieniacza jonowego nie jest dokładnie takie samo, jak w przypadku zastosowanego do równoważenia kolumny buforu. Wynika to z tak zwanego efektu Donnana, który powoduje, że protony są przyciągane lub odpychane, w zależności od ładunku nośnika, w jego mikrośrodowisku. Na ogół w pobliżu nośnika pH jest do 1jednostki wyższe niż to w otaczającym buforze dla wymienników anionowych i 1 jednostkę niższe dla wymienników kationów. W konsekwencji, jeżeli białko jest adsorbowane na żywicy kationowymiennej przy pH 5, to będzie narażone na pH 4, i jeśli ma słabą stabilność przy tej wartości pH, to może ulegać denaturacji.
Tabela 2. Przykłady jonowymieniaczy stosowanych w chromatografii.

Jak już wspomniano, pH wyjściowego buforu powinno wynosić co najmniej jedną jednostkę pH powyżej albo poniżej pI białka w celu zapewnienia odpowiedniego wiązania do jonowymieniacza. Należy jednak pamiętać, że jonowymieniacze takie jak CM i DEAE są przykładami słabych wymienników jonowych. Słaby jonowymieniacz ulega jonizacji tylko w ograniczonym zakresie pH. Tak więc DEAE zaczyna „tracić” ładunek przy pH powyżej 9, podczas gdy CM zaczyna „tracić” ładunek poniżej pH ~5. Termin „słaby” nie odnosi się do siły wiązania jonów do żywicy ani fizycznejwytrzymałości samego złoża. Skuteczny, wyjściowy zakres pH przy zastosowaniu DEAE lub CM zawiera się w granicach pH 5 - 9. Korzystając z jonowymieniaczy należy zwrócić uwagę na matrycę, do której jest on przyłączony (Sepharose - Agaroza; Sephadex – Dekstran; Sephacel - Celuloza).
Rodzaj wypełnienia ma także wpływ na stabilność jonowymieniacza (Tabela 3).
Tabela 3. Przykładowe parametry złóż zawierających jonowymieniacz DEAE.

Przy sile jonowej w przedziale od zera do wartości fizjologicznych (0,15–0,2 M) niektóre białka wykazują tendencję do tworzenia precypitatów na skutek niewystarczających ładunków powodujących odpychanie się cząsteczek białka. Natomiast, gdy stężenie soli jest za wysokie, wówczas białka ulegają wysalaniu. Oprócz elucji w gradiencie soli, białka można wymywać zmieniając pH buforu, co powoduje zmianę stopnia jonizacji bocznych reszt aminokwasów, a co za tym idzie ładunku białka. Należy jednak pamiętać, aby w danym pH białko pozostawało stabilne, oraz o tym, że białka wypadają z roztworu w przy pH równym ich pI. Dla tych białek, wymywanie w gradiencie pH może być niemożliwe (Rysunek 4).
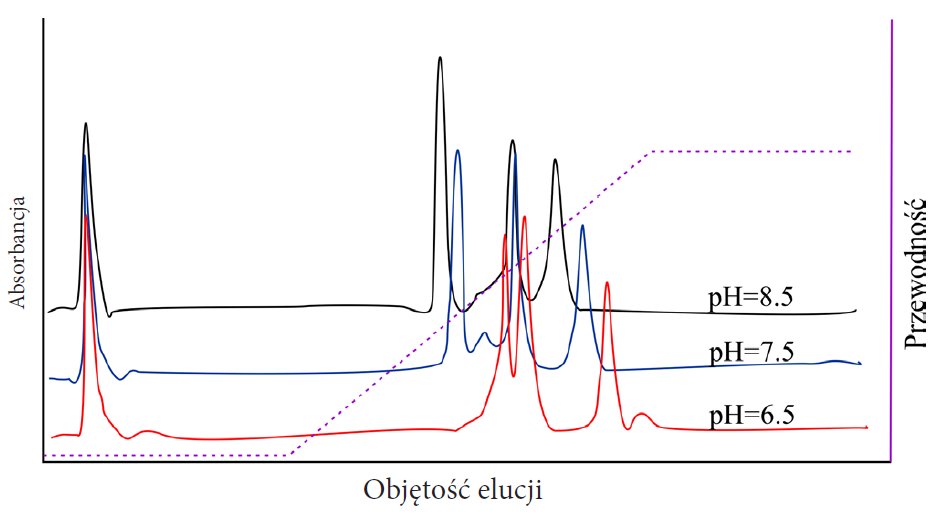
Rysunek 4. Zmiana rozdzielczości rozdziału metodą chromatografii IEC pod wpływem pH (wg. http://www.bio-rad.com/en-pl/applications-technologies/liquid-chromatography-principles/ion-exchange-chromatography).
Ważnym parametrem wpływającym na rozdzielenie analitów z wykorzystaniem chromatografii jonowymiennej jest wielkość ziaren wypełnienia do którego przyłączona jest grupa funkcyjna jonowymieniacza. Wraz ze zmniejszaniem się wielkości ziaren matrycy rośnie rozdzielczość, jednakże dobra selektywność (stopień separacji między pikami chromatograficznymi) jest ważniejszym czynnikiem niż wysoka rozdzielczość i zależy nie tylko od charakteru i ilości grup funkcyjnych na powierzchni matrycy, ale także od warunków doświadczalnych takich jak pH (wpływają na ładunek białka), siła jonowa i warunki elucji.
Chromatografia oddziaływań hydrofobowych
Ważną techniką oczyszczania białek, wykorzystującą ich hydrofobową naturę, jest chromatografia oddziaływań hydrofobowych (HIC). Rozdział w chromatografii HIC uwarunkowany jest różnicami w sile oddziaływania obszarów hydrofobowych białka z bardziej hydrofobowymi grupami ligandów przyłączonymi do nośnika pozbawionego ładunku elektrycznego. Technikę HIC można z powodzeniem wykorzystać po rozdziale metodą IEC, ponieważ rozdzielaną próbkę białka umieszcza się w buforze o wysokim stężeniu soli. W celu elucji białek z kolumny HIC stosuje się malejący gradient soli (Rysunek 5). Technika HIC to nieinwazyjna metoda chromatograficzna o minimalnych możliwościach degradacji rozdzielanych biomolekuł. Związane jest to ze stabilizującym działaniem soli i słabymi oddziaływaniami z matrycą. Mimo to, technika ta zapewnia bardzo dobre parametry oczyszczania.

Rysunek 5. Schemat oczyszczania białek za pomoc techniki HIC wraz z przykładowym chromatogramem.
Za oddziaływania hydrofobowe odpowiadają aminokwasy takie jak izoleucyna, walina, leucyna i fenyloalanina. Aminokwasy te nie tworzą wiązań wodorowych z wodą. Z tego powodu, najczęściej znajdują się w hydrofobowym rdzeniu natywnego białka lub w części lipidowej membrany. Ponieważ tylko niewielką część aminokwasów można „schować” niektóre hydrofobowe aminokwasy znajdują się na powierzchni białka. Hydrofobowość natywnych białek jest zatem sumą hydrofobowości eksponowanych łańcuchów bocznych i części szkieletu białkowego. Hydrofobowa adsorpcja białek jest procesem termodynamicznie korzystnym ze względu na entropię, a siłą napędową tego procesu jest ograniczenie powierzchni styku z wodą.
Najczęściej stosowane ligandy w chromatografii oddziaływań hydrofobowych to łańcuchy alkilowe terminalnie modyfikowane grupami aminowymi. Wykorzystywanymi ligandami są również łańcuchy alkilowe wykazujące silny charakter hydrofobowy (n-butyl, n-oktyl) lub aromatyczne grupy arylowe, np. fenyl. Ligandy arylowe mają mieszane własności, tj. oddziałują zarówno przez liniowe fragmenty łańcucha, jak i wykorzystują oddziaływania typu π−π pomiędzy aromatycznymi pierścieniami grup arylowych.
Grupa fenylowa ma w przybliżeniu tę samą hydrofobowość jak grupa pentylowa, chociaż ligand fenylowy może mieć bardzo różną selektywność w porównaniu do liganda pentylowego. Przy stałej gęstości powierzchniowej liganda, zdolność złoża do wiązania białek wzrasta wraz z długością łańcuchów alkilowych, jednakże zbyt długie łańcuchy alkilowe mogą utrudnić resorpcję białek o dużej hydrofobowości. Najczęściej stosowane są ligandy o długości łańcucha wynoszącej ~4-10 atomów węgla. Dłuższe łańcuchy wykorzystywane są w przypadku białek błonowych.
Nie ma ogólnych zasad wyboru liganda (alkilowy czy arylowy). Dobór odpowiednich nośników zależy od warunków rozdzielania białek i jest dobierany eksperymentalnie. W związku z tym nie można przenosić warunków wiązania i elucji z jednego typu liganda na inny.
W chromatografii HIC rodzaj i stężenie soli bardzo silnie wpływają na oddziaływanie białko-ligand.
Ze wzrostem stężenia soli rośnie ilość białka wiązanego z hydrofobowymi ligandami. Na początku obserwowany jest wzrost liniowy, jednakże po przekroczeniu pewnego stężenia ilość wiązanego białka wzrasta wykładniczo. Wysolenie białka w kolumnie może niekorzystnie wpływać na selektywność rozdzielania chromatograficznego.
Nie tylko stężenie soli, ale także jej rodzaj, ma wpływ na parametry rozdziału. Poniżej przedstawiono które aniony i kationy faworyzują powstawanie oddziaływań hydrofobowych.
Aniony: PO43- > SO42- > CH3COO- > Cl- > Br- > NO3- > ClO4- > I- > SCN-
Kationy: NH4+ > Rb+ > K+ > Na+ > Li+ > Mg2+ > Ca2+ > Ba2+
Przekładając to na szereg soli (Na2SO4 > K2SO4 > (NH4)2SO4 > Na2HPO4 > NaCl > KCl > LiCl > NaBr > KBr) można zaobserwować, że najlepsze z nich to siarczany sodu, potasu i amonu, jednak nie zawsze korzystne jest ich stosowanie. (NH4)2SO4 jest niestabilny i może powodować wydzielanie się amoniaku, dlatego należy stosować pH >8. Natomiast zbyt wysokie stężenie Na2SO4 może powodować problem z rozpuszczalnością białek.
Innymi czynnikami, które należy uwzględniać w przypadku chromatografii HIC jest stężenie jonów wodorowych (wartość pH) i temperatura. Zaobserwowano, że siła oddziaływań hydrofobowych białka z hydrofobowym ligandem jest tym większa, im niższa jest wartość pH.
Z drugiej strony, podwyższenie wartości pH umożliwia redukcję wielopunktowych oddziaływań hydrofobowych pomiędzy makromolekułą a długimi łańcuchami alifatycznymi, gęsto rozmieszczonymi na powierzchni nośnika. Wartość pH powyżej 9-10 sprzyja wzrostowi hydrofilowości co utrudnia wiązanie do nośników.
Temperatura odgrywa istotną rolę we właściwym przeprowadzeniu rozdziału HIC ponieważ oddziaływania hydrofobowe makromolekuł z hydrofobowym ligandem mają charakter sił van der Waalsa, które są zależne od temperatury. Im wyższa temperatura, tym oddziaływania hydrofobowe są silniejsze. Stosowanie wysokiej temperatury nie zawsze jest korzystne i może doprowadzić do denaturacji białka.
Oddziaływania hydrofobowe pomiędzy złożem a białkami można wykorzystać nie tylko w chromatografii HIC. Inną metodą wykorzystującą te oddziaływania jest chromatografia w warunkach odwróconego układu faz lub w układzie faz odwróconych (RPC). Mimo pewnych podobieństw pomiędzy tymi technikami powierzchnia wypełnienia w odwróconych układach faz jest zazwyczaj bardziej hydrofobowa niż w przypadku chromatografii HIC. Prowadzi to do silniejszych oddziaływań pomiędzy złożem a białkami. W celu skutecznej elucji związanych białek lub peptydów trzeba wykorzystać bardziej niepolarne rozpuszczalniki organiczne jak acetonitryl lub metanol. W chromatografii RP wiązanie ligandu jest funkcją podziału fazowego pomiędzy hydrofobowe własności rozpuszczalnika, a grupy funkcyjne na kolumnie. Podczas korzystania z tej techniki białka ulegają w mniejszym lub większym stopniu denaturacji, co wynika z hydrofobowej natury całej sekwencji polipeptydowej a nie tylko pojedynczych aminokwasów (HIC). Ponieważ większość aminokwasów hydrofobowych znajduje się wewnątrz białka globularnego, wiązanie do złoża zależy od stopnia denaturacji białka i dostępności tych grup dla ligandów kolumny. W przypadku HIC stopień podstawienia ligandów wynosi średnio 10-50 µmoli (C2-C8 arylo) ligandów na cm3 matrycy, a w chromatografii RPC stosuje się kilkaset µmoli (C4-C8 alkilo) ligandów na cm3 matrycy. Wielkość ziaren matrycy wynosi od 2-20 µm, przy czym złoża o małej wielkości ziaren stosuje się do rozdziału aminokwasów i nukleotydów, a o dużej do rozdziału białek. Podsumowując, peptydy oczyszczane są na złożach o średnicy ziaren rzędu 5-15 µm.
Przez długie lata RPC wykorzystywano w układach HPLC. Stworzenie złóż typu SOURCE (Source 15RPC i Source 30RPC), opartych na matrycy zbudowanej z polistyrenu sieciowanego diwinylobenzenem pozwoliło uzyskać niezwykłą stabilność i odporność mechaniczną złoża przy znacznie obniżonym oporze przepływu i bardzo niskiej wartości ciśnienia zwrotnego w kolumnie. Dzięki tym rozwiązaniom udało się wprowadzić technikę RPC w zakresie niskich i średnich ciśnień przy utrzymaniu pożądanych parametrów selektywności i rozdzielczości metody stosowanej dotychczas w chromatografii HPLC.
Chromatografia RPC oferuje dużą elastyczność warunków separacji. Dzięki tej technice możliwe jest uzyskanie wysokiej rozdzielczości pomimo niewielkich różnic w hydrofobowości poszczególnych białek. Rozdział mieszaniny białek może być prowadzony zarówno metodą izokratycznej elucji jak i elucji gradientowej, szczególnie jeśli istotne jest skrócenie czasu analizy. Ponadto, chromatografię RPC można wykorzystać do odsalania preparatu białek i zatężania hydrofobowych składników próbki, co w połączeniu z możliwością używania rozpuszczalników organicznych sprawia, że RPC jest techniką wykorzystywaną w połączeniu ze spektrometrią mas.
Kolejną techniką chromatograficzną jest sączenie molekularne, tzw. filtracja żelowa (Gel filtration Chromatography, GFC). Metoda ta pozwala na rozdzielanie białek na podstawie ich wielkości i kształtu. W przeciwieństwie do chromatografii adsorpcyjnej, w której wiązanie biomolekuł następuje już na początku kolumny co prowadzi do zagęszczania próbki w GFC warunki rozdziału powodują rozcieńczenie próbki. Podstawowym etapem przed zadozowaniem próbki do układu chromatograficznego jest jej odpowiednie zagęszczenie. Dodatkowym ograniczeniem jest stężenie białka, które ma zostać rozdzielane. Przykładowo, etap rozdzielania może być zrealizowany z wykorzystaniem kolumny Superdex 75 (10/300 GL). W przypadku tego złoża, dla uzyskania dobrej separacji stężenie białek nie może przekraczać 10 mg . mL-1 a objętość próbki powinna wynosić od 200 do 500 µL. Maksymalnie rozdzieleniu może podlegać około 5 mg białka całkowitego. Ze względu na budowę materiału tworzącego złoże GFC ograniczeniu podlega także szybkość rozdziału chromatograficznego. Dla wspomnianej powyżej kolumny wynosi ona 0,5 - 1 mL . min-1. GFC jest również techniką, która znacznie wydłuża czas analizy w przypadku stosowania systemów niskociśnieniowych.
Oczywiście należy wspomnieć o ważnych zaletach tej metody. Rozdzielanie może być przeprowadzane w obecności niezbędnych jonów, kofaktorów, detergentów, w buforze o wysokiej lub niskiej sile jonowej; w temperaturze 37 °C lub w zimnym pomieszczeniu, zgodnie z wymaganiami eksperymentu.
Oczyszczone białka można rozdzielać w dowolnie wybranym buforze (niezależnie od buforu, w którym znajduje się nasze białko), a ponadto można dokonać wymiany buforu lub odsolenia próbki (Sephadex G-25). Współczesne sita molekularne używane w GFC odporne są także na działanie 8M mocznika lub 6M chlorowodorku guanidyny. Sephacryl i Superdex są na ogół bardziej odporne na działanie soli chaotropowych lub skrajnych wartości pH niż klasyczne złoża, takie jak Sepharose™ lub Sephadex.
W przeciwieństwie do innych rodzajów nośników selektywność matrycy w GFC nie jest regulowana przez zmianę składu fazy ruchomej. W optymalnych warunkach nie występuje adsorpcja do złoża a faza ruchoma powinna być traktowana jako faza nośna, bez wpływu na rozdział chromatograficzny. W przypadku chromatografii żelowej cząsteczki biomolekuł nie są adsorbowane, lecz opóźniany jest ich przepływ przez kolumnę, przez to ten typ rozdziału kwalifikowany jest jako izokratyczny. Schemat rozdzielania analitów w przypadku chromatografii żelowej przedstawiono na rysunku 6.
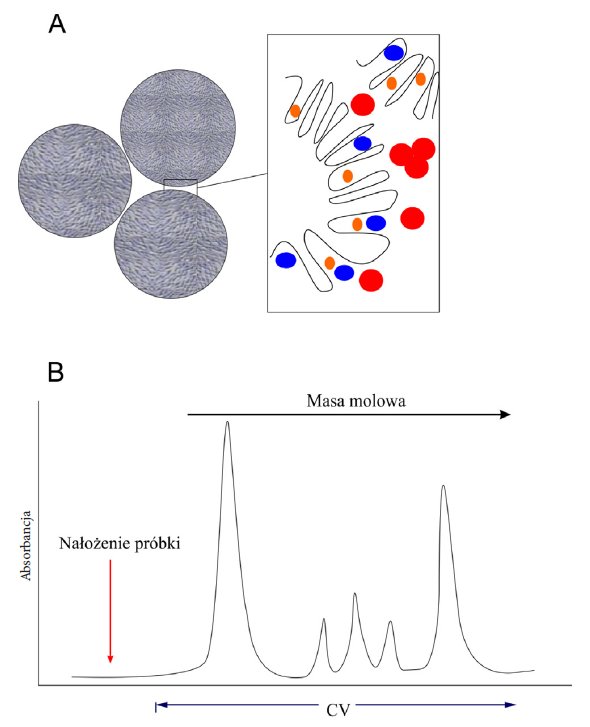
Rysunek 6. Chromatografia GFC. (A) Schematyczne przedstawienie ziaren złoża (B) Przykładowy chromatogram rozdzielania związków z wykorzystaniem chromatografii żelowej. Szerokość pasm wskazuje na stopień rozcieńczenia (im większa tym większe rozcieńczenie próbki).
Podczas rozdzielania małych cząsteczek, takich jak jony i małe białka, następuje dyfuzja w głąb ziaren złoża dzięki czemu poruszają się one wolniej w kolumnie. Cząsteczki większe lub o wydłużonym kształcie wypływaj z kolumny jako pierwsze (Rysunek 6), inaczej mówiąc rozdzielanie poszczególnych białek następuje na skutek dyfuzji różnicowej. Złoże zbudowane jest zwykle z porowatych kulek (z porami o określonym rozkładzie wielkości) i obojętnego, wysoko uwodnionego żelu. Do produkcji złóż wykorzystuje się dekstran, agarozę oraz żel poliakrylamidowy. Złoża do GFC produkowane są o różnym stopniu porowatości, dzięki czemu frakcjonować można mieszaniny białek o różnych masach molowych, peptydy czy kwasy nukleinowe.
Chromatografia GFC mimo łatwości przeprowadzenia wrażliwa jest na wiele czynników. Jednym z nich jest charakterystyka nanoszonej próbki. Parametry na jakie należy zwrócić uwagę w przypadku etapu przygotowania próbek do analizy z wykorzystaniem chromatografii GFC przedstawiono w Tabeli 4.
Tabela 4. Przygotowanie próbek do chromatografii GFC.
Tabela 4. Przygotowanie próbek do chromatografii GFC.
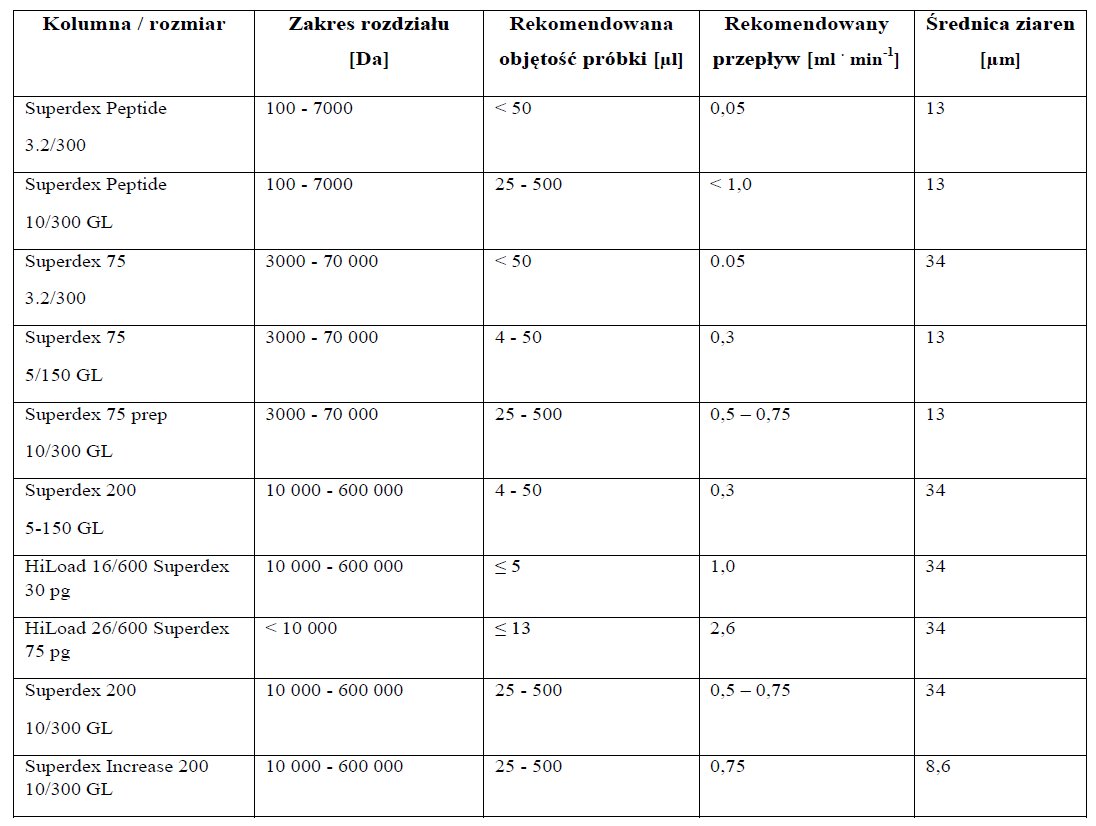
Mając dobrze przygotowaną próbkę do iniekcji należy zastanowić się nad wyborem kolumny. Obecnie na rynku jest duży wybór kolumn (Tabela 5).
Tabela 5. Przykład kolumn stosowanych w GFC.
Należy tylko wybrać odpowiednią dla planowanej aplikacji.
Przy wyborze kolumny istotna jest objętość dozowanej próbki, tak aby była możliwie mała oraz aby możliwości rozdzielcze złoża były odpowiednie dla konkretnego białka (Rysunek 7). Dla próbki A lepszą kolumną będzie Superdex Peptide niż Superdex 75, pomimo iż obie kolumny zapewniają rozdzielczość w zakresie masy molowej tej próbki. Podobnie w przypadku próbki B (lepsza jest kolumna Superdex 200). Nie tylko kolumny oparte na złożu Superdex zapewniają dobre parametry rozdzielania. Innym złożem jest Sephacryl HR (o dużej rozdzielczości). Sephacryl zapewnia rozdział, w zależności od średnicy ziaren, w zakresie mas molowych od 102 do 108.
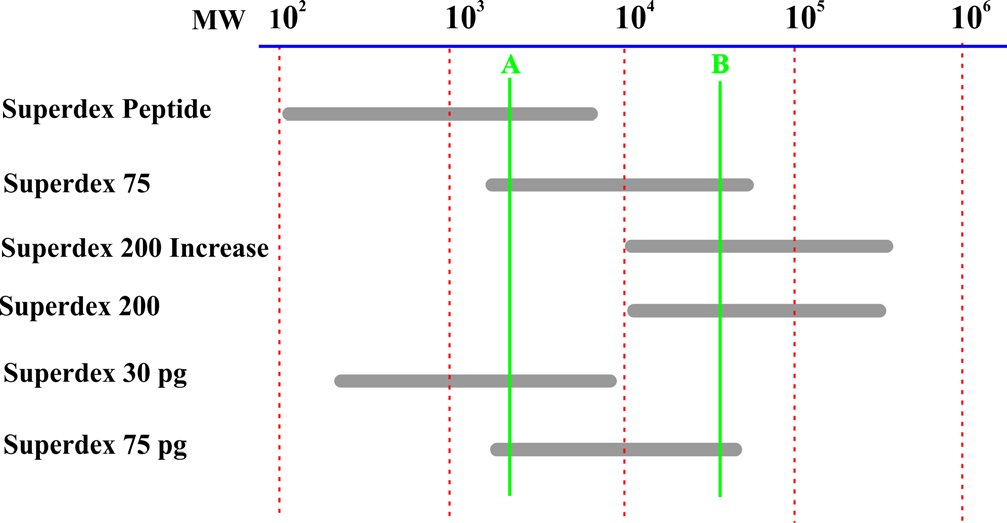
Rysunek 7. Zakresy rozdzielczości poszczególnych kolumn chromatograficznych.
Jak już wspomniano, GFC wydaje się łatwą techniką separacyjną, jednakże obarczona jest dużym prawdopodobieństwem popełnienia błędów, poprzez np. nieodpowiednią objętość próbki czy nieodpowiednią lepkość. Dobranie rozdzielczości złoża zależy także od stopnia zrównoważenia kolumny, długości kolumny, ciśnienia w kolumnie i szybkości rozdziału. W przypadku kolumn do GFC obserwuje się oddziaływania hydrofobowe złoża z rozdzielanymi białkami. Należy wtedy obniżyć stężenie soli lub zastosować dodatek 5 % izopropanolu. Korzystne efekty przynosi także zmiana pH, szczególnie jeśli istnieje możliwość występowania oddziaływań jonowych. Niewłaściwie dobrany bufor i jego parametry mogą także doprowadzić do wytrącenia się białek w kolumnie, dlatego przed rozdzielaniem należy dokładnie sprawdzić czy białko nie wytrąca się w buforze, w którym będzie prowadzony proces rozdzielania. Można także zastosować rozdział w warunkach redukujących. Dokładny opis wszystkich problemów oraz jak im przeciwdziałać można znaleźć na stronach internetowych producentów kolumn.
Na Rysunku 8 przedstawiono sytuacje w których można mówić o bardzo dobrym rozdziale bądź słabym rozdziale.
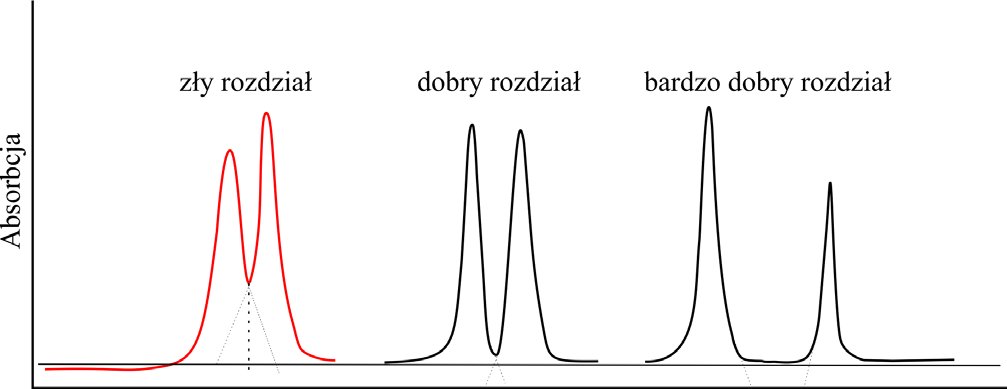
Rysunek 8. Jakość rozdziału w chromatografii GFC.
Szczególnym typem złoża stosowanym w chromatografii żelowej jest Sephadex LH20.
Sephadex LH-20 wytwarza się poprzez hydroksypropylowanie Sephadexu G-25, usieciowanej matrycy zbudowanej z dekstranu. Złoże to zostało opracowane specjalnie do filtracji żelowej produktów naturalnych, takich jak steroidy, witaminy, terpeny, lipidy i peptydy o niskiej masie cząsteczkowej (do 35 aa). Przeznaczone jest głównie do zastosowania w rozpuszczalnikach organicznych, choć może być także wykorzystywane do rozdziału w roztworach wodnych. Sephadex LH-20 z powodzeniem używany jest w celach analitycznych jak i preparatywnych. Dużą zaletą tego złoża jest wysoka selektywność oraz wydajność związana z podwójnym charakterem matrycy (hydrofilowym i lipofilowym) oraz bardzo dobra powtarzalność rozdziałów.
Chromatografia GFC często wykorzystywana jest do odsalania preparatów białek. Najczęściej w tym celu wykorzystuje się kolumny zawierające Sephadex G-25 w układzie pojedynczym lub wielokrotnym, co zwiększa wydajność odsolenia.
Chromatografia powinowactwa
Biologiczna funkcja białek często związana jest z ich zdolnością do specyficznych interakcji z innymi cząsteczkami, zwanymi ligandami. Oddziaływania te mogą następować pomiędzy białkami a substancjami o niskiej masie cząsteczkowej, takimi jak substraty, kofaktory lub inhibitory, ale w szczególności interakcje te występują pomiędzy białkami. Interakcje te występują pomiędzy białkiem, a dokładnie jego komplementarną częścią i pasującym do niego ligandem. Oddziaływania te mogą obejmować oddziaływania elektrostatyczne, hydrofobowe, mogą także występować na poziomie wiązań van der Waalsa oraz wiązań wodorowych. Chromatografia AC (Affinity Chromatography) umożliwia rozdzielanie białek na podstawie odwracalnej reakcji dwóch wykazujących specyficzne powinowactwo substancji, z których jedna – ligand, związana jest ze stałym nośnikiem.
W technice AC specyficzny ligand jest kowalencyjnie przyłączony do obojętnej matrycy chromatograficznej (Rysunek 9). Niekorzystne przyłączenie ligandu do matrycy może powodować zakłócanie zdolności do wiązania z cząsteczką docelową. Aby wyeliminować ten problem pomiędzy matrycę a ligand wprowadza się element dystansowy. Dzięki temu ligand znajduje się w pewnej odległości od powierzchni matrycy, co zmniejsza sferyczną przeszkodę wiązania, która może występować, gdy ligand związany jest bezpośrednio z matrycą. Elementy dystansowe odgrywają bardzo ważną rolę w przypadku małych unieruchomionych ligandów, rzadko stosuje się je dla ligandów makrocząsteczkowych. Optymalna długość pomiędzy matrycą a ligandem wynosi od 6 do 10 atomów węgla lub ich odpowiedników. Oczywiście wiązanie matryca-ligand musi być fizykochemicznie obojętne.
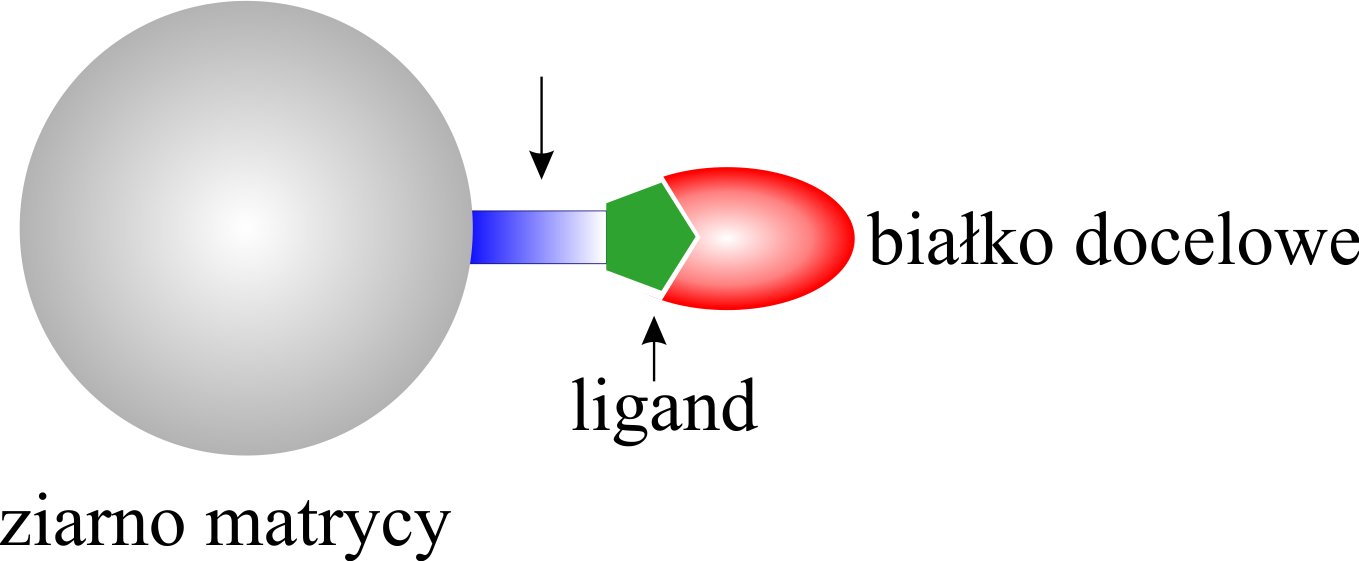
Rysunek 9. Schemat wiązania pomiędzy układem matryca-ligand a docelowym białkiem.
Dzięki chromatografii AC możliwe jest oczyszczanie próbek, tworzących układy enzym-substrat, enzym-inhibitor, przeciwciało-antygen, hormon-receptor, kwasy nukleinowe-białka. W chromatografii AC próbka dozowana jest w warunkach, które szczególnie i odwracalnie sprzyjają wiązaniu białka docelowego. Ponieważ tylko pożądane białka są adsorbowane z ekstraktu, pozostałe białka przechodzą przez kolumnę lub są łatwo wypłukiwane. Do elucji cząsteczki docelowej stosuje się zmienione warunki dzięki czemu oddziaływanie białko-ligand ulega osłabieniu.
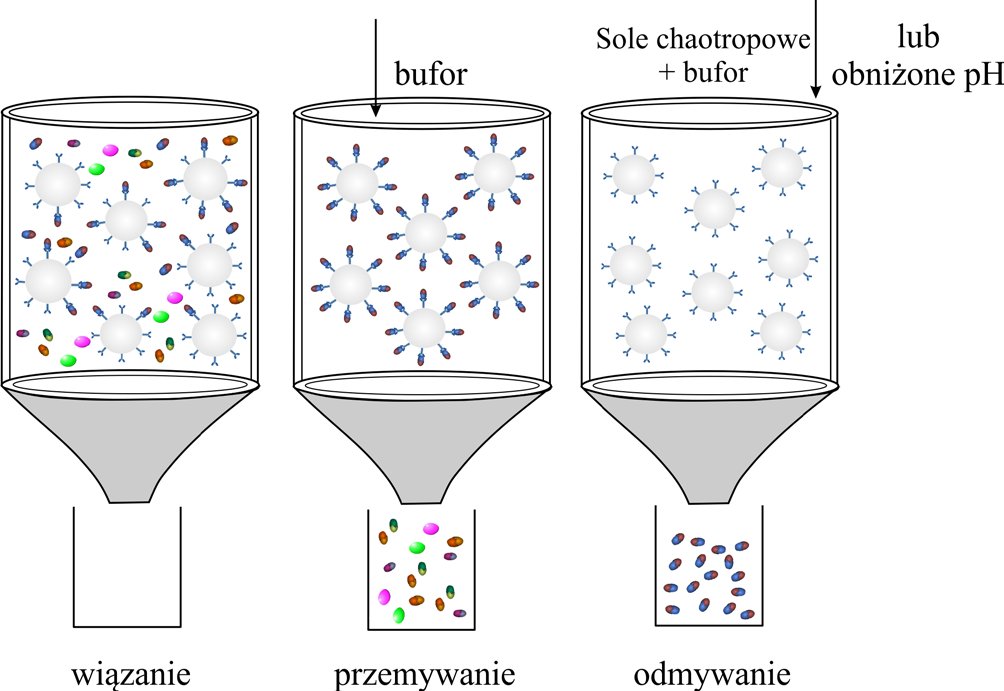
Rysunek 10. Schemat oczyszczania białek za pomocą techniki chromatografii powinowactwa na przykładzie immunopowinowactwa.
Występowanie specyficznych reakcji jest bardzo pożądane, jednakże nie jest to regułą i czasem także inne białka wiążą się z ligandem.
W chromatografii powinowactwa ważne są nie tylko przyłączone ligandy, ale także sama matryca. Idealny materiał żelowy powinien charakteryzować się odpowiednimi cechami. Po pierwsze, musi on posiadać odpowiednie grupy chemiczne, do których ligandy mogą być kowalencyjnie przyłączone i posiadać względnie duże pole powierzchni dostępne do przyłączenia. Powinien być obojętny na działanie rozpuszczalników i buforów, które stosuje się w procesie przyłączania ligandów oraz oczyszczania - zwłaszcza podczas elucji białka. Najbardziej przydatne są hydrofilowe i obojętne matryce, które jednocześnie umożliwiają oddziaływanie dużych białek z ligandem a nie matrycą, i nie blokują swobodnego przepływu przez złoże. Do produkcji odpowiednich matryc wykorzystuje się szereg syntetycznych, organicznych i nieorganicznych, porowatych substancji, na przykład usieciowany dekstran, polistyren, celulozę, poliakrylamid, porowate szkło i krzemionkę.
Złoża dostępne komercyjnie często pozbawione są opisu badań dotyczących metod przyłączania ligandów do złoża, co może stanowić ciekawy problem badawczy.
Wydajność i jakość oczyszczonych biomolekuł zależy nie tylko od matrycy i ligandów ale w dużej mierze od warunków wiązania, przemywania i uwalniania ze złoża. Najkorzystniejsza jest sytuacja gdy zoptymalizowane warunki wiązania gwarantują, że cząsteczki docelowe mogą skutecznie oddziaływać z ligandem i są zatrzymywane a niespecyficzne interakcje są zminimalizowane. W większości przypadków bufor wiążący stosuje się również do przemywania złoża i odmywania substancji niezwiązanych z kolumny. Fundamentalne znaczenie ma szybkość przepływu przez kolumnę. Jeśli wymuszony przepływ jest zbyt wysoki, może nie dochodzić do prawidłowego wiązania.
Czasem zamiast gotowej kolumny bardziej korzystne jest zakupienie samego złoża i pustej kolumny, którą samodzielnie można upakować. W przypadku powinowactwa jest to proces o nieznacznym stopniu skomplikowania. Dzięki takiemu postępowaniu możliwe jest wydłużenia czasu wiązania białka z ligandem nawet do kilkunastu godzin. Poza tym dzięki powolnemu wiązaniu przy delikatnym mieszaniu złoża zwiększa się wydajność wiązania ligandów z docelowym białkiem niwelując wpływ przeszkód sterycznych i zanieczyszczeń.
Kolejnym etapem jest przemywanie złoża, podczas którego istotny jest odpowiedni dobór parametrów buforu, którego się używa. Najczęściej stosuje się ten sam bufor, w którym nanosi preparat, jednakże podczas wiązania ze złożem często dochodzi do niespecyficznych połączeń pomiędzy ligandem, złożem, białkiem docelowym i białkami zanieczyszczającymi. W tym wypadku konieczne jest odpowiednie dobranie warunków przemywania np. poprzez zwiększenie stężenie soli lub obniżenie pH buforu. W ostatnim z etapów ważne jest takie dobranie warunków aby podczas procesu uwalniania związanego białka nastąpiła dysocjacja kompleksu ligand-białko, i aby nie dochodziło do zniszczenia aktywności białka (jeśli to enzym) oraz degradacji złoża. Dobranie odpowiednich warunków podczas tego etapu jest czynnikiem generującym najwięcej trudności w przypadku chromatografii powinowactwa. Istotne jest również odpowiednie zoptymalizowanie czasu uwalniania. Proces ten musi następować na tyle szybko aby nie dochodziło do zbyt dużego rozcieńczenia próbki zawierającej oczyszczane białko.
Jak już wspomniano, interakcje ligand-białko są często oparte na kombinacji oddziaływań elektrostatycznych, hydrofobowych czy wiązaniach wodorowych. W takim przypadku stosuje się niespecyficzne eluenty. Staranne rozpatrzenie względnego znaczenia tych trzech typów interakcji dla stabilności białka związanego może pomóc w doborze odpowiedniego eluentu.
Zmiana pH eluentu wpływa na jonizację grup ligandu i cząsteczki docelowej. W związku z tym najczęściej stosowanym sposobem wymywania substancji silnie związanych niespecyficznie jest obniżenie pH buforu. Często wystarczające jest obniżenie wartości pH o około 2 jednostki. Zdarza się, że osiągane są lepsze rezultaty, gdy pH wzrasta (stosowanie wodorotlenku amonu). Jeśli wymywanie osiągane jest poprzez zmianę wartości pH, to bardzo często koniecznym staje się neutralizowanie pH zebranych frakcji bezpośrednio po elucji w celu zminimalizowania ryzyka denaturacji białka. W przypadku oddziaływań elektrostatycznych zwiększa się siłę jonową stosując 1 M rozwór NaCl.
Gdy wiązanie jest zdominowane przez silne oddziaływania hydrofobowe, należy zastosować bardziej drastyczne warunki np. stosując sól chaotropową lub środek denaturujący. Ten typ wymywania często wykorzystuje się w przypadku immunoabsorpcji z wykorzystaniem unieruchomionych przeciwciał poliklonalnych.
Jedną z najskuteczniejszych technik oczyszczania białek jest chromatografia immunopowinowactwa, wykorzystująca unieruchomione przeciwciała. Wysoka swoistość przeciwciał pozwala je zakwalifikować jako niezwykle użyteczne w szczególności gdy nie znamy innego ligandu komplementarnego z naszym białkiem. Przez wiele lat stosowano jako immunoadsorbent przeciwciała poliklonalne, które są mieszaniną przeciwciał o różnej specyficzności i wiążących różne epitopy antygenu. Pomimo, że są łatwe do otrzymania, to ze względu na duże zróżnicowanie w ich specyficzności istnieje niebezpieczeństwo wystąpienia reakcji krzyżowej, co w konsekwencji może uniemożliwić skuteczne oczyszczanie danego białka.
Obecnie, coraz częściej stosuje się adsorbent na bazie przeciwciał monoklonalnych. Nowoczesna technika hybrydomy i odpowiednie sposoby badań przesiewowych dają możliwość uzyskania przeciwciał o praktycznie dowolnej swoistości i powinowactwie, jednak wadą przeciwciał monoklonalnych nadal pozostaje ich wysoki koszt.
Z powodu bardzo silnego wiązania między przeciwciałem a jego antygenem (Kd od l0-8 do l0-12 M) często oczyszczanie wymaga surowych warunków elucji (8 M mocznik lub 6 M chlorowodorek guanidyny). Może to doprowadzić do inaktywacji białka docelowego lub nawet zniszczenia części ligandu. Użyteczne może być zastosowanie środków chaotropowych (3 M KSCN) i obniżenie pH do około 3. Wadą immunopowinowactwa jest także fakt, że niektóre z przeciwciał często wymywa się z kolumny podczas oczyszczania i muszą być one usuwane z eluatu docelowego białka.
Czasami chromatografia powinowactwa stosowana jest w przeciwnym układzie i antygenami są białka stosowane jako ligandy w chromatografii AC. Takie podejście umożliwia oczyszczanie specyficznego przeciwciała. W tym celu powszechnie wykorzystywane jest białko A lub białko G. Ligandy te są białkami błony komórkowej drobnoustrojów z rodzaju Staphylococcus lub Streptococcus posiadającymi wysokie powinowactwo do regionu stałego IgG. Wiązanie jest zwykle osiągane przy fizjologicznych wartościach pH a niższe wartości pH (około 3) wykorzystuje się do wymywania.
W chromatografii AC często stosuje się grupę białek zwanych lektynami, które wykazują zdolność do specyficznego oddziaływania i odwracalnego wiązania się do węglowodanów lub grupy węglowodanów. Unieruchomione lektyny są bezcennym narzędziem do oddzielania glikokoniugatów takich jak glikoproteiny, polisacharydy, glikolipidy, a nawet całych komórek zawierających glikoproteiny o specyficznych strukturach węglowodanowych na błonie komórkowej. Najszerzej stosowane są lektyny z roślin strączkowych ze względu na ich liczebność, np. konkanawalina A, która oddziałuje silnie z mannozą i glukozą.
Inną grupą białek stosowanych w AC są białka wiążące DNA. Stanowią one bardzo zróżnicowaną klasę białek, a ich zdolność do wiązania kwasów nukleinowych wykorzystywana jest do ich oczyszczania. W tym celu jako ligand stosuje się siarczan heparyny. Heparyna jest glikozaminoglikanem, imituje polianionową strukturę kwasu nukleinowego i w ten sposób oddziałuje silnie z białkami wiążącymi DNA. Oczywiście białka wiążące DNA mogą być izolowane za pomocą odpowiedniej sekwencji DNA jako ligandu. W ten sposób izolowane są czynniki transkrypcyjne.
W celu izolacji enzymów stosuje się ligandy mogące być substratem, odwracalnym inhibitorem konkurencyjnym lub allosterycznym aktywatorem. Na przykład monofosforan adenozyny (AMP) może być wykorzystany do wiązania białek wykazujących powinowactwo do AMP, ADP i ATP. Para-aminobenzamidyna często używana jest jako ligand do oczyszczania proteaz serynowych i esteraz, np. trypsyny, trombiny, kalikreiny, urokinazy itd.
Chociaż istnieje kilka systemów powinowactwa to dla większości białek docelowych brakuje odpowiedniego ligandu nadającego się do wychwytywania na stałym podłożu. W celu wyeliminowania tej przeszkody wykorzystuje się modyfikacje genetyczne polegające na fuzji genu kodującego białko docelowe z genem kodującym białko o odpowiednim powinowactwie. Kiedy białko chimeryczne ulega ekspresji znacznik umożliwia specyficzny wychwyt białka fuzyjnego. System oczyszczania białek fuzyjnych może być oparty na wielu rodzajach interakcji. Przykładami powszechnie używanych znaczników są GST-tag (S-transferaza glutationowa), białko wiążące maltozę (MBP), znacznik FLAG, S-tag, peptyd wiążący kalmodulinę, Bio-tag, Strep-tag oraz His-tag (6 histydyn).
Jedną z odmian chromatografii powinowactwa jest metalopowinowactwo. Jednakże ze względu na to, że technika ta opiera się na tworzeniu relatywnie słabych wiązań koordynacyjnych pomiędzy jonami metalu a niektórymi aminokwasami w białku, powinno mówić się raczej o technice pseudopowinowactwa. Jednym z aminokwasów szczególnie odpowiednim do wiązania jonów metali jest histydyna, która umożliwia wytworzenie najsilniejszych oddziaływań. Histydyna jako donor elektronów w pierścieniu imidazolowym łatwo tworzy wiązania koordynacyjne z unieruchomionym metalem przejściowym. Cysteina umożliwia wiązanie jonów metali poprzez wolną grupę sulfhydrylową (warunki redukcyjne). Aromatyczne łańcuchy boczne tryptofanu, fenyloalaniny i tyrozyny także mogą oddziaływać z jonami metali lecz w praktyce oczyszczanie białek opiera się przede wszystkim na dostępności reszt histydyny. Ważnym czynnikiem wiązania do jonów metali jest występowanie reszt histydyny na powierzchni białka i ich odpowiednia ilość w sekwencji tak, aby powstawały odpowiednio silne wiązania koordynacyjne.
Jako chelatory jonów metali przyłączone do złoża agarozowego wykorzystuje się: iminodioctan (IDA), kwas nitrylotrioctowy (NTA) oraz tris(karboksymetylo)-etylenodiaminę (TED). Rysunek 11 przedstawia schemat związanego do złoża chelatora z przyłączonym jonem metalu. Istotne jest, aby chelator nie wykorzystywał wszystkich możliwych do wytworzenia wiązań koordynacyjnych.

Rysunek 11. Schemat związanego do złoża chelatora z przyłączonym jonem metalu.
Pozorne powinowactwo białka do metalu zależy w dużym stopniu od rodzaju jonu metalu. Najbardziej powszechnie wykorzystywane są jony dwuwartościowych metali przejściowych: Fe2+, Co2+, Ni2+, Cu2+ i Zn2+. Możliwe jest także wykorzystanie jonów trójwartościowych takich jak: Fe3+ i Al3+. Siła wiązania pomiędzy białkiem a jonem metalu zależy od chelatora i samego jonu. W przypadku chelatora IDA powinowactwo białek do jonów metali przedstawia się następująco: Cu2+ > Ni2+ > Zn2+ > Co2+.
Praca ze złożami do chromatografii powinowactwa wymaga zachowania odpowiednich warunków. Bufory powinny być pozbawione substancji redukujących oraz innych chelatorów jak np. EDTA. Ponadto należy unikać buforów o pH kwaśnym (poniżej 5), gdyż powodują uwalnianie jonów metali. Może to prowadzić do oksydacyjnego uszkodzenia białek. Najkorzystniejsze jest przeprowadzenie wiązania w pH obojętnym lub lekko zasadowym i zwiększonej sile jonowej (0,1-1 M NaCl) a wymywanie prowadzić roztworem imidazolu lub EDTA. Jeśli uwolnione białko będzie dalej analizowane np. metodą dichroizmu kołowego należy pozbyć się imidazolu lub uwalniać białka buforem o niskiej kwasowości.
Podsumowując, w porównaniu do innych metod AC chromatografia powinowactwa do jonów metali jest niespecyficzna, mało powtarzalna, zaś jony metali mogą być ługowane, a do złoża często wiążą się inne białka niż docelowe. Zaletą są niskie koszty, łatwa regeneracja złoża, możliwość oczyszczania w warunkach denaturujących, szczególnie dla białek ekspresjonowanych w E. coli i otrzymywanych w postaci ciałek inkluzyjnych.
Większość przedstawionych powyżej informacji dotyczy chromatografii niskociśnieniowej. Nie można zapominać jednak o wysokosprawnej chromatografii cieczowej (HPLC). Omówienie HPLC przekracza jednak zakres tego artykułu.


